

LA CLASSIFICAZIONE DELLE MALATTIE MITOCONDRIALI
La classificazione delle malattie mitocondriali riflette una caratteristica peculiare del sistema
della fosforilazione ossidativa, quella di essere costituita da proteine codificate da due diversi sistemi genetici,
il genoma mitocondriale e il genoma nucleare.
Un primo gruppo di malattie è caratterizzato dalla presenza di mutazioni del mtDNA, a trasmissione materna o
a insorgenza sporadica. Un secondo gruppo è causato da mutazioni in geni nucleari che fanno parte o controllano
la fosforilazione ossidativa (OXPHOS) e la corretta funzionalità mitocondriale. Queste malattie sono a volte
classificate sulla base delle sole alterazioni biochimiche rilevate dall'analisi dei tessuti affetti
(soprattutto muscolo scheletrico), perché i geni responsabili ancora non si conoscono. L'enorme eterogeneità clinica,
biochimica e molecolare di queste patologie spiega perché solo il 50% circa delle malattie mitocondriali definite
sul piano clinico-biochimico, sono diagnosticate geneticamente.
Negli ultimi anni molti progressi sono stati compiuti in questo campo grazie all'introduzione di nuove tecnologie
(Next generation sequencing NGS) che offrono la possibilità di analizzare rapidamente e a basso costo
un gran numero di sequenze di DNA. Questo ha portato all'identificazione di numerosi geni nucleari
associati a patologie mitocondriali e di conseguenza ha permesso di ampliare e migliorare l'offerta
diagnostica a pazienti e famiglie.
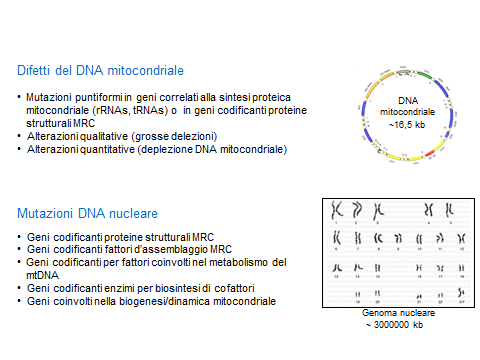
1. Mutazioni del mtDNA
A seconda delle caratteristiche molecolari e genetiche delle mutazioni del mtDNA, questo gruppo di difetti
comprende sindromi causate da mutazioni puntiformi del mtDNA,
dovute a riarrangiamenti su larga scala del mtDNA o legate a riduzione della quantità di mtDNA.
1.1 Mutazioni puntiformi del mtDNA
Si tratta di quadri clinici associati a sostituzioni di singole basi o a micro-inserzioni/micro-delezioni, nella molecola del mtDNA. Queste mutazioni possono interessare sequenze codificanti RNA transfer (tRNA), RNA ribosomiali (rRNA), o RNA messaggeri (mRNA) di proteine codificate dal mtDNA. Quasi tutte le mutazioni puntiformi sono trasmesse per via matrilineare. Spesso, ma non sempre, queste mutazioni sono eteroplasmiche. Sebbene, ad oggi, centinaia di mutazioni puntiformi siano state descritte in associazione con uno spettro molto eterogeneo di presentazioni cliniche, le mutazioni di gran lunga più frequenti sono poche, e sono associate a sindromi cliniche piuttosto ben definite.
La neuropatia ottica ereditaria di Leber (Leber's Hereditary Optic Neuropathy, LHON) (OMIM535000), a esordio in età giovanile e netta prevalenza nel sesso maschile, è caratterizzata dalla perdita acuta o subacuta della visione centrale con esito in atrofia ottica. Il deficit visivo, che a parte rari casi, è permanente, costituisce in genere l'unica manifestazione della malattia: raramente sono presenti anche alterazioni del ritmo cardiaco (sindrome di pre-eccitazione ventricolare). La biopsia muscolare non evidenzia la presenza di fibre ragged-red, e solitamente la biopsia non è in questo caso necessaria per la diagnosi. La malattia è stata associata a mutazioni nelle posizioni nucleotidiche m.3460, 11778 e 14484, che interessano rispettivamente le subunità ND1, ND4 e ND6 del complesso I della catena respiratoria. Altre mutazioni, sempre in geni del complesso I, sono state identificate. Molti aspetti della sindrome LHON rimangono da chiarire, compresi l'estrema specificità tissutale delle lesioni cliniche, la prevalenza nel sesso maschile, e le conseguenze biochimiche di ciascuna mutazione.
La sindrome caratterizzata da neuropatia, atassia e retinite pigmentosa (Neurogenic muscle weakness, ataxia, retinitis pigmentosa, NARP) (OMIM551500) può comprendere, oltre ai suddetti segni, epilessia e decadimento mentale. La malattia ha in genere esordio in età adulta. Anche in questo caso, la biopsia muscolare non mostra la presenza delle tipiche fibre ragged red. La malattia è stata associata alla mutazione m. 8993T>G, nella sequenza codificante la subunità 6 dell'ATPasi mitocondriale (complesso V della catena respiratoria). Nella stessa posizione è inoltre descritta una transizione T>C presente in pazienti con fenotipo NARP meno grave. La stessa mutazione m. 8993T>G, presente con un grado di eteroplasmia nettamente più elevato (>90%) rispetto al fenotipo NARP, è stata anche riscontrata in piccoli pazienti affetti da sindrome di Leigh (v. oltre) senza deficit biochimico di citocromo c ossidasi o piruvico deidrogenasi.
La Encefalomiopatia Mitocondriale con Acidosi Lattica ed episodi simil-Stroke (Mitochondrial Encephalomyopathy, Lactic Acidosis and Strokelike episodes, MELAS) (OMIM540000) è definita dalla presenza delle seguenti manifestazioni: 1) episodi di tipo ictale (stroke-like) causati da lesioni cerebrali focali spesso localizzate nelle aree parieto-occipitali; 2) acidosi lattica o comunque livelli anormali di lattato nel sangue (e liquor); 3) fibre "ragged-red" nella biopsia muscolare. Altri segni di coinvolgimento del sistema nervoso centrale comprendono deterioramento mentale, cefalea ricorrente con vomito "cerebrale", epilessia focale o generalizzata, e sordità neurosensoriale. La malattia è trasmessa per via materna e l'esordio è variabile, dalla primissima infanzia all'età giovanile-adulta.
La sindrome MELAS è tipicamente associata alla mutazione m.3243A>G, nel gene codificante il tRNA per la Leucina (codone UUR). Sono state in seguito riportate altre mutazioni puntiformi associate a MELAS, anche se si tratta di casi più rari.
La mioclono-epilessia con fibre ragged-red (Myoclonus Epilepsy with Ragged Red Fibers, MERRF) (OMIM545000) è caratterizzata dall'associazione di mioclono, epilessia, debolezza e ipotrofia muscolare, atassia , e, talvolta, deterioramento mentale. L'entità delle manifestazioni cliniche può essere estremamente variabile nell'ambito della stessa famiglia. Tale variabilità si ritiene sia in relazione alla quantità di mtDNA mutato rispetto al normale (eteroplasmia) e alla variabilità nella distribuzione tissutale della mutazione. La maggior parte delle famiglie affette è portatrice della transizione m.8344A>G, nel gene codificante il tRNA per la lisina.
Numerose altre mutazioni puntiformi del mtDNA sono state associate a diversi fenotipi clinici in singoli pazienti o in poche famiglie.
1.2 Alterazioni qualitative del mtDNA
Si può trattare di delezioni parziali del mtDNA o, più raramente, di duplicazioni parziali. Entrambi i tipi di mutazione sono eteroplasmici, dato che coesistono sempre con una quota di mtDNA normale, e sono per lo più sporadici. Queste alterazioni grossolane del mtDNA sono quasi invariabilmente associate con tre principali presentazioni cliniche: la sindrome di Kearns-Sayre, la sindrome di Pearson e l'Oftalmoplegia Esterna Progressiva.
La sindrome di Kearns-Sayre (KSS) è una grave malattia ad insorgenza sporadica caratterizzata dalla triade: 1) Oftalmoplegia Esterna Progressiva (PEO) con ptosi (abbassamento) palpebrale bilaterale; 2) Retinopatia Pigmentaria; 3) insorgenza prima dei 20 anni. Segni aggiuntivi frequenti sono l'incoordinazione motoria (atassia) di origine cerebellare, il deterioramento mentale, la sordità, e alterazioni del ritmo cardiaco. Vi è spesso ritardo della crescita.
La sindrome di Pearson è una rara malattia sporadica della primissima infanzia caratterizzata da anemia sideroblastica, pancitopenia e insufficienza del pancreas esocrino con malassorbimento intestinale. Nei bambini che sopravvivono oltre i primi anni, si assiste a un progressivo miglioramento della situazione ematologica e gastrointestinale, mentre insorgono i segni caratteristici della sindrome di Kearns-Sayre.
La Oftalmoplegia Esterna Progressiva (PEO) è caratterizzata dall'insorgenza in età adulta di ptosi bilaterale e paralisi dei movimenti oculari, spesso associate a debolezza dei muscoli delle spalle e del bacino di grado variabile. E' spesso associata alla presenza di delezioni multiple a carico del mtDNA. Mutazioni in geni nucleari sono state associate a forme di PEO a trasmissione dominante o recessiva (vedi capitolo "mitocondriopatie dovute a mutazioni in geni nucleari").
1.3 Alterazioni quantitative del mtDNA
Una riduzione della quantità del mtDNA è comunemente chiamata "deplezione". Questa alterazione si associa comunemente a delle forme a insorgenza infantile ad andamento progressivo. Gli organi più spesso coinvolti sono: muscolatura scheletrica e cardiaca, fegato e cervello. Le deplezioni del mtDNA sono causate da mutazioni in geni nucleari (vedi capitolo "mitocondriopatie dovute a mutazioni in geni nucleari").
2. Mitocondriopatie dovute a mutazioni in geni nucleari
Oltre il 90% delle proteine mitocondriali sono espressione di geni nucleari e comprendono la maggior parte dei componenti del sistema OXPHOS, le proteine necessarie per l'assemblaggio dei complessi della catena respiratoria. Inoltre la replicazione del mtDNA, la sua trascrizione e traduzione sono dipendenti da proteine a codifica nucleare.
Un numero crescente di malattie degenerative ereditarie, soprattutto in campo neurologico o neuromuscolare, si è rivelato essere dovute a mutazioni in geni codificanti proteine con localizzazione mitocondriale, più o meno direttamente correlate alla OXPHOS.
Accenniamo qui di seguito a una sola malattia, la Sindrome di Leigh, perché si tratta della malattia più nota e frequente del gruppo dei disordini "nucleari" dei mitocondri (anche se, come riportato nel paragrafo 1.1 circa il 20% dei casi è dovuto a mutazioni del mtDNA, per lo più nel gene dell'ATPasi 6 e più raramente a carico di geni codificanti le subunità della COX o in geni tRNA). La sindrome di Leigh è l'encefalopatia mitocondriale più frequente nell'infanzia. Essa è primariamente un'entità neuropatologica- neuroradiologica, caratterizzata da lesioni focali bilaterali in una o più aree della sostanza grigia profonda a livello dei nuclei della base, del midollo spinale rostrale, del tronco encefalo, dei talami e del cervelletto. I sintomi clinici sono correlati alle aree coinvolte nel processo di necrosi cellulare, inizialmente consistono in ipotonia e regressione psicomotoria, seguite dalla comparsa in tempi variabili di distonia, movimenti involontari, atassia e tetraparesi spastica. Quasi costantemente si associano sintomi sistemici con difficoltà ad alimentarsi, vomiti frequenti e scarso accrescimento staturo-ponderale, probabilmente in relazione allo stato di acidosi metabolica. Anche il sistema nervoso periferico è frequentemente affetto per la presenza di polineuropatia assonale e demielinizzante. Un'alterazione del metabolismo energetico mitocondriale è suggerita dal frequente aumento dei livelli di acido lattico nel siero e nel liquor. In circa il 30% dei casi, il deficit biochimico principale è costituito da un difetto importante dell'attività del complesso IV (citocromo c ossidasi) e l'alterazione genetica è in questo caso dovuta, nella maggior parte dei casi, a mutazioni in un gene "assemblatore" del complesso IV, chiamato SURF1. In altri casi il difetto biochimico è a carico del complesso I o del complesso II della catena respiratoria, e mutazioni delle subunità di questi complessi sono state in effetti identificate in alcuni pazienti. In alcuni casi di s. di Leigh è stato riscontrato un deficit di Piruvico Deidrogenasi, associato a mutazioni del gene legato al cromosoma X codificante la subunità E1-alfa dell'enzima.
Per classificare le mitocondriopatie dovute a mutazioni nei geni nucleari può essere utilizzata una suddivisione dei prodotti proteici in base al loro ruolo biologico :
Ci sono alcune proteine che non sono comprese in questa classificazione ma sono indirettamente correlate alla OXPHOS e le cui mutazioni possono causare malattie mitocondriali: ad es. le proteine necessarie per l'import delle proteine all'interno del mitocondrio (TIMM8A, DNAJC19), proteine legate all'apoptosi (AIFM1), proteine detossificanti molecole dannose per i mitocondri (ETHE1). Queste condizioni sono estremamente rare.
2.1 Malattie dovute ad alterazioni dei componenti della catena respiratoria
Sebbene 72 delle 85 subunità del sistema OXPHOS sono codificate dal DNA nucleare, mutazioni in questi geni sono molto rare. Questo suggerisce che siano estremamente dannose e probabilmente embrio-letali. Le mutazioni descritte sono solitamente associate a esordio precoce o neonatale. L'analisi dei geni codificanti le subunità OXPHOS non è però stata effettuata in passato in maniera sistematica, soprattutto per il complesso I. Grazie all'introduzione di tecniche NGS è in crescita il numero di segnalazioni riguardanti mutazioni in questi geni: la maggior parte sono alterazioni a carico del complesso I descritte in pazienti a esordio infantile, anche se sono state riportate mutazioni nelle subunità strutturali dei complessi II, III, IV e V. Le presentazioni cliniche più frequenti sono: sindrome di Leigh, leucoencefalopatie e cardiomiopatie.
2.2 Malattie dovute a mutazioni nei fattori di assemblaggio dei complessi della catena respiratoria
I complessi del sistema OXPHOS sono multieteromerici e per il loro corretto assemblaggio è necessario l'intervento di diversi fattori; mutazioni a carico di questi fattori portano alla produzione di complessi instabili o solo parzialmente funzionanti. Per i difetti a carico dei complessi III, IV e V, alterazioni genetiche in geni di questa categoria sono molto più frequenti rispetto a quelle a carico delle subunità strutturali riportate nel paragrafo precedente. Le principali presentazioni cliniche sono le stesse, con vari tipi di encefalocardiomiopatie.
2.3 Malattie dovute a mutazioni in proteine che controllano il metabolismo del mtDNA
Il mtDNA dipende dal genoma nucleare in quanto questultimo codifica proteine coinvolte nella replicazione, trascrizione, traduzione, riparo e stabilità del mtDNA.
La replicazione del mtDNA richiede un piccolo set di proteine: la DNA polimerasi gamma (POLG), l'elicasi Twinkle (PEO1), la mitochondrial single-stranded DNA binding protein (mtSSB); e il rifornimento di deossinucleotidi trifosfato (dNTPs). Difetti in questi enzimi sono spesso associati a delezioni del mtDNA (alterazioni qualitative; vedi paragrafo 1.2), mentre i difetti a carico del pool di dNTPs solitamente portano a deplezione del mtDNA (alterazioni quantitative; vedi paragrafo 1.3). Oftalmoplegia Esterna Progressiva e debolezza muscolare sono tipiche dei geni che causano alterazioni qualitative mentre i geni associati ad alterazioni quantitative (deplezioni) causano diverse malattie con coinvolgimento neurologico, muscolare ed epatico.
La traduzione del mtDNA o sintesi proteica mitocondriale avviene all'interno dei mitocondri ad opera di un sistema composto da tRNA e rRNA sintetizzati in situ a partire dai corrispondenti geni mitocondriali e da proteine codificate dal DNA nucleare e importate all'interno dei mitocondri. Diverse mutazioni a carico di subunità dei ribosomi mitocondriali, di enzimi che intervengono nella maturazione dei tRNA mitocondriali, di fattori di elongazione, in aminoacil tRNA sintetasi sono state identificate in pazienti con malattie mitocondriali e rappresentano uno dei gruppi clinicamente più eterogenei.
2.4 Malattie dovute a mutazioni in enzimi coinvolti nella biosintesi di cofattori
Il coenzima Q10 o ubichinone (CoQ) è un componente lipofilico della catena di trasporto degli elettroni, che trasferisce elettroni dai complessi I e II al complesso III. Il CoQ ha anche un ruolo come antiossidante e come stabilizzatore di membrana. Mutazioni a carico degli enzimi responsabili della biosintesi del CoQ sono solitamente associate a un deficit di CoQ a livello muscolare e possono causare mioglobinuria e atassia a esordio precoce.
I complessi della catena respiratoria sono localizzati nella membrana mitocondriale interna, composta prevalentemente da cardiolipina. Questa sostanza ha un ruolo essenziale per il corretto funzionamento del sistema OXPHOS, quindi alterazioni nella biosintesi della cardiolipina causano malattie mitocondriali (in particolare la sindrome di Barth, una cardiopatia dovuta a mutazioni nel gene TAZ o tafazzina).
I complessi OXPHOS richiedono cofattori come rame, gruppi eme o centri ferro-zolfo (Fe-S) per svolgere la loro funzione di trasporto di elettroni. Un ampio gruppo di proteine, fattori di assemblaggio ed enzimi coinvolti nella biosintesi e nell'incorporazione di gruppi prostetici è necessario per avere complessi enzimatici funzionali. Le presentazioni fenotipiche associate a queste disfunzioni sono molto varie: miopatia, atassia, leucoencefalopatia, anemia sideroblastica
2.5 Malattie dovute a mutazioni in proteine coinvolte nella biogenesi/dinamica dei mitocondri
I mitocondri non sono organelli statici e isolati ma formano una complessa rete interconnessa. I meccanismi di fusione e fissione mitocondriale richiedono apparati proteici a livello delle membrane mitocondriali interna ed esterna. La proteina principale per la regolazione della morfologia della membrana interna è OPA1, le cui mutazioni sono causa di atrofia ottica. Neuropatie tipo Charcot-Marie-Tooth sono invece associate a mutazioni in MFN2, proteina di fusione della membrana esterna mentre mutazioni in geni codificanti componenti del sistema di fissione mitocondriali (DNM1L, MFF) sono solitamente responsabili di gravi encefalopatie ad esordio precoce.

Mappa mutazionale del mtDNA.